科研进展丨材料计算与数据库平台在单原子催化领域取得进展
近日,松山湖材料实验室/中国科学院物理研究所孟胜研究员和刘淼副研究员团队,在国际权威期刊《The Journal of Physical Chemistry Letters》上发表了题为“Orbital Dependence in Single-Atom Electrocatalytic Reactions”的研究论文,提出催化性能主要取决于参与反应的部分d亚层轨道,解决了d带理论和前沿轨道理论一直以来的争议。值得一提的是,该论文还被编辑推荐作为封面文章发表。


在催化领域中,由Hammer和Nørskov在1995年提出的d带理论被证明是用来预测过渡金属催化活性最好用的理论之一。d带理论指出,过渡金属表面催化剂的催化活性主要与过渡金属的d能带相关,且d带中心是催化活性的有效描述符。然而,近些年越来越多的研究工作表明,d带理论在很多情况下是不成立的,于是就有了2020年Wu等人提出的前沿轨道理论。前沿轨道理论指出,过渡金属单原子催化剂的催化活性与费米面附近前沿轨道的空间结构和方向息息相关。因此,d带理论和前沿轨道理论就产生了很大的分歧。
松山湖材料实验室材料计算与数据库平台的孟胜研究员和刘淼副研究员团队,通过密度泛函理论计算方法,对N掺杂石墨烯负载过渡金属单原子催化剂(TM−N−C)体系进行了全面系统的研究。结果表明,在催化反应中只有dz2轨道参与了反应,且dz2轨道中心是催化活性的有效描述符,并且单个原子的微小局部环境可以显著地调节催化反应活性。也就是说,d带理论可以推广到d亚层轨道水平,晶格应变或过渡金属原子z向位移引起的晶体场的微小扰动,可以显著地改变与反应相关的d亚层轨道,进而影响催化活性。有趣的是,参与反应的d亚层轨道同时也是前沿轨道,也就表明前沿轨道理论和d带理论的结论是一致的。该工作成功地将d带理论和前沿轨道理论统一了起来,为人们理解过渡金属催化的微观物理机制提供理论参考。
松山湖材料实验室材料计算与数据库平台博士后王亚南为论文第一作者,松山湖材料实验室/中国科学院物理研究所孟胜研究员和刘淼副研究员为论文通讯作者。该研究成果得到了国家重点研发计划基金(2021YFA0718700)、中国科学院基金(CAS-WX2021PY-0102, ZDBS-LY-SLH007, XDB33020000)、广东省基础与应用基础研究基金(2020A1515110743)、中国博士后科学基金(2019TQ0347)等多个项目的大力支持。
相关研究结果于2022年6月23日在线发表在《The Journal of Physical Chemistry Letters》上。文章链接:https://doi.org/10.1021/acs.jpclett.2c01381。
文章附件:JPCL.pdf
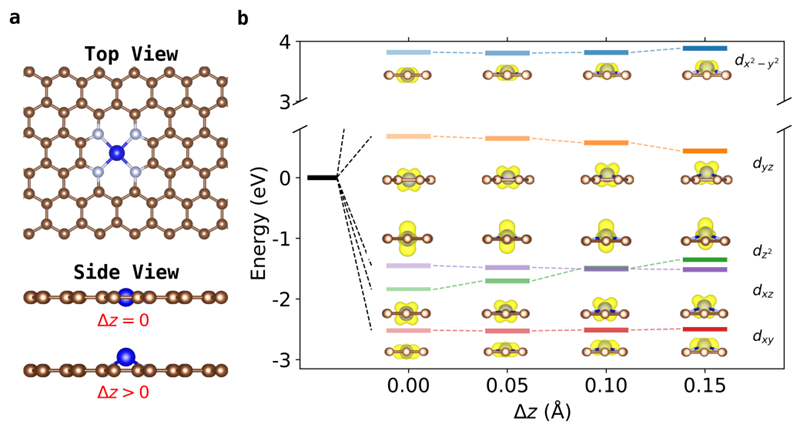
图1(a)TM−N−C单原子催化剂的原子结构图。(b)TM−N−C在晶体场下的d能带劈裂
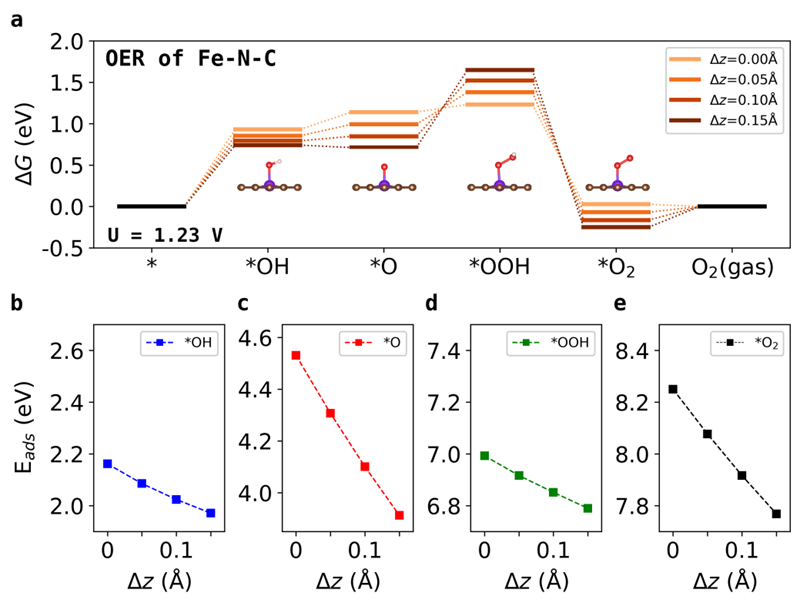
图2(a)当Fe−N−C的Z方向位移从0增加到0.15 Å时,OER过程的自由能变化图谱。
(b-c)Fe−N−C体系OER过程中的*OH,*O,*OOH,和*O2四个中间态的吸附能随z方向位移的变化情况。
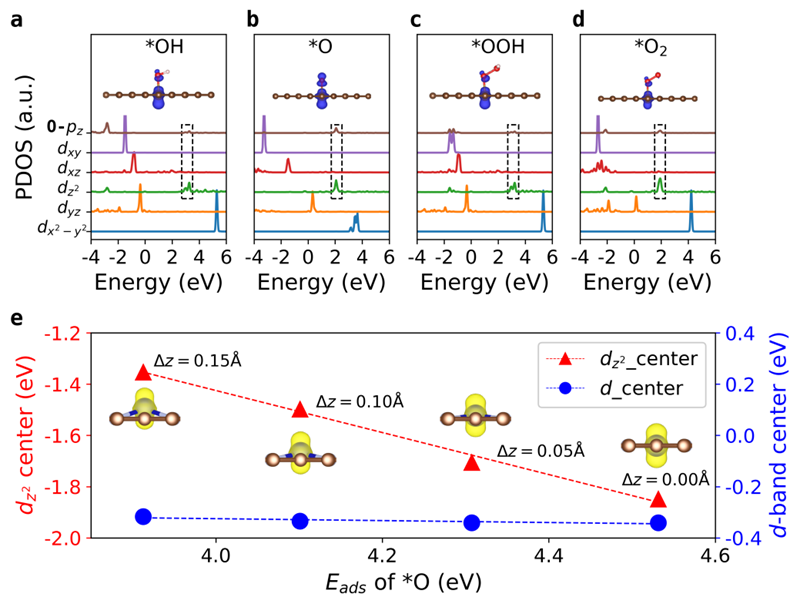
图3 (a-d)Fe−N−C体系OER过程的*OH、*O、*OOH和*O2四个中间态的电子态密度和轨道杂化图。
(e)当Fe−N−C的z方向位移从0增加到0.15 Å时,O原子的吸附能与Fe原子的dz2轨道中心和d带中心的相关性。
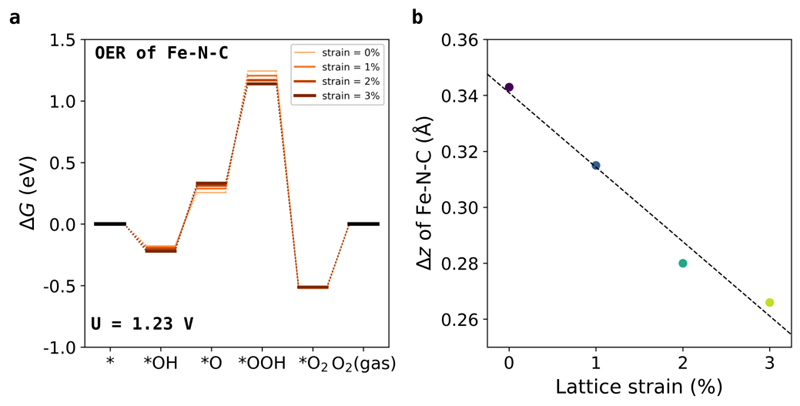
图4 (a)当晶格应变从0增大到3%时,Fe−N−C体系OER过程的自由能变化图谱。
(b)Fe−N−C体系OER过程的*O中间态中,晶格应变与z方向位移的关系。