科研进展丨“人工智能+大数据”开启材料物性的高精度预测
“人工智能+大数据”变革性地改进了诸多行业的生产力,也为材料科学的进步带来新的机遇。近日,松山湖材料实验室/中国科学院物理研究所孟胜研究员和刘淼副研究员团队,在《SCIENCE CHINA Materials》上发表了题为“A universal model for accurately predicting the formation energy of inorganic compounds”的研究论文,该工作依托超大规模的第一性原理计算材料数据集,构建出无机晶体形成能的高精度预测泛化模型,为新材料搜索提供了一种高效、低成本的结合能预测手段。

随着数据科学和材料科学的进步,人们如今可以构建出较为准确的人工智能模型,用于材料各种性质的预测。然而,人工智能模型的预测精度和泛化本领通常不可得兼。此前业内的绝大多数无机晶体形成能模型,往往要经过大量的数据清洗。因此,虽然这些模型的精度很高,但是泛化本领非常差,应用价值不高。材料计算与数据平台依托实验室内一流的基础设施,及自研材料数据库(https://atomly.net/),以其中170,714个无机晶体化合物的高通量第一性原理计算数据集为基础,训练得到了可精确预测无机化合物形成能的机器学习模型。相比于同类工作,该项研究以超大数据集为出发点,构建出无机晶体形成能的高精度泛化模型,可外推至广阔相空间,其中的DenseNet神经网络模型精度可以达到R2 = 0.982和平均绝对误差(MAE) = 0.072 eV atom−1。上述模型精度的提升源自一系列新型特征描述符的构建,这些描述符可有效提取出原子与邻域原子间的电负性和局域结构等信息,从而精确捕捉到原子间的相互作用。本文为新材料搜索提供了一种高效、低成本的结合能预测手段。
从物理思维出发,本项工作提出了基于元素电负性之差的化合物形成能的特征描述符,使人工智能模型的预测精度得到了大幅提升。同时,后续的随机森林模型进一步验证了元素电负性之差与化合物形成能的强相关性,印证了物理思维的正确性,也为提升人工智能模型的可解释性提供了新思路。论文结尾展示了该模型对于部分体系的验证结果,并精准的预测了若干离子化合物体系的形成能。该模型可部分替代目前的量子力学计算任务,将小时级的计算作业缩减为秒级计算,具有实际应用价值。借助模型,平台将进一步探索更广阔的无机材料相空间,为材料科学的发展开辟新的大陆。
松山湖材料实验室材料计算与数据库平台博士后梁英宗为论文第一作者,松山湖材料实验室/中国科学院物理研究所孟胜研究员和刘淼副研究员为论文通讯作者。该研究成果得到了松山湖材料实验室和中国科学院(CAS-WX2021PY-0102, ZDBS-LY-SLH007, XDB33020000)的支持。
相关研究结果于2022年7月27日在线发表在《SCIENCE CHINA Materials》上。文章链接:https://www.sciengine.com/SCMs/doi/10.1007/s40843-022-2134-3
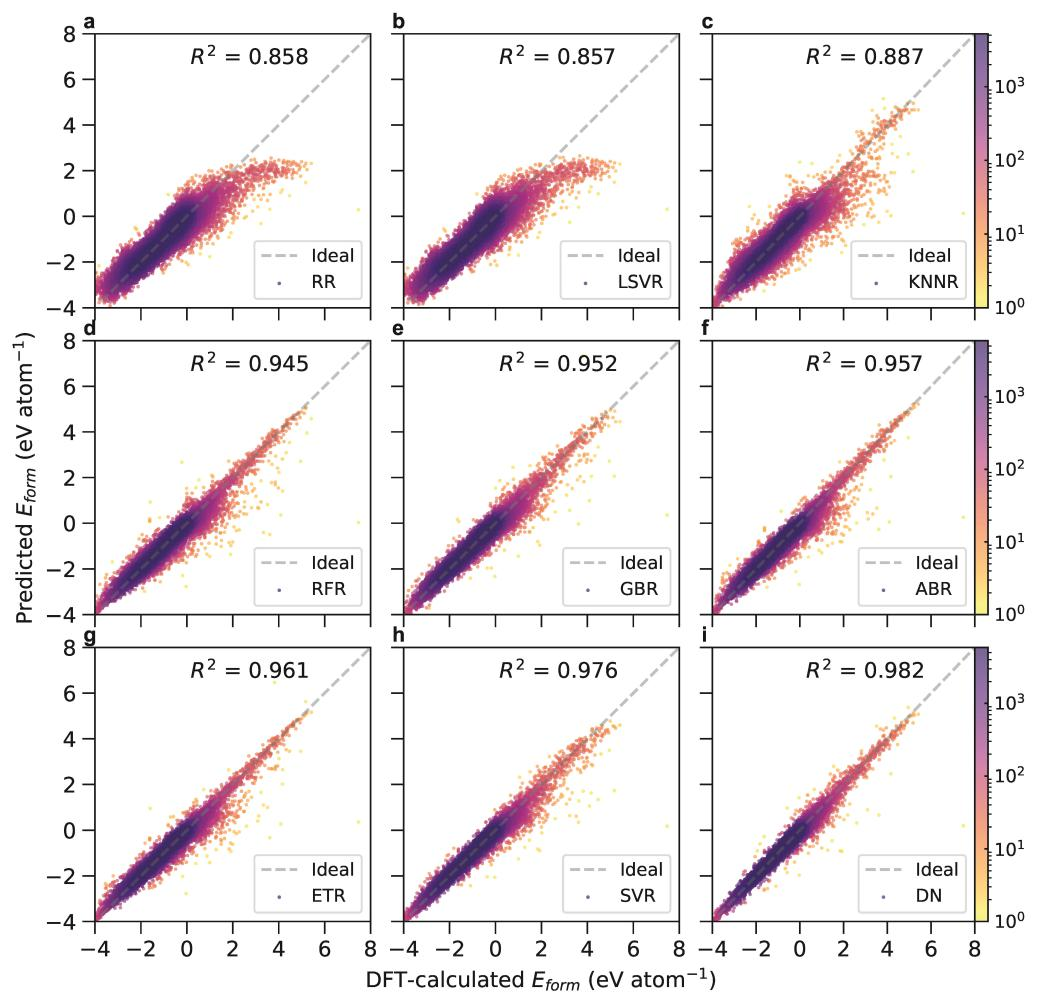
图1. 人工智能模型vs量子力学计算。图为不同的机器学习算法的预测精度,其中图(i)中的DenseNet模型达到R2 = 0.982 和MAE = 0.072 eV atom−1。
.png)
图2. (a)Ti-O,(b)V-O,(c)Mn-O,和(d)Li-P 化学系统的热力学相图。蓝色三角形为通过量子力学计算得到的形成能,红色圆形为模型预测数值。可见模型可较准确的预测量子力学计算结果。
原文附件:/SCM_2022.pdf